

Executive Summary
With decentralized clinical trials (DCTs), some or all aspects of the clinical trial are conducted in locations that are not centralized at a specific clinical or research site. DCTs are trials executed either in whole or in part remotely, through telemedicine, mobile technologies, local sites, and mobile healthcare providers. This may greatly increase the efficiency and/or reach of a trial, but there are specific differences between a DCT and a traditional trial that an Institutional Review Board (IRB) or Ethics Committee (EC) should consider. This guide is intended to summarize these considerations to help investigators, sponsors, and IRB/ECs understand these differences in order to implement the appropriate level of oversight of DCTs. The recommendations contained herein are based on discussions of a multi-stakeholder task force of experts representing academia, IRBs, patients, regulators, trial sponsors, and sites. The Multi-Regional Clinical Trials (MRCT) Center of Harvard and Medable, as the initiative co-hosts, used the discussions to create resources to support IRB/EC review of clinical trials with decentralized elements.
As shown in Figure 1, the primary concerns are to ensure the rights and welfare of the participants (people), the integrity and security of remote data collection (data collection), and the adequacy of trial and data oversight (remote data oversight). Each of the 12 potential DCT elements falls within one of these domains. In addition, several cross-cutting themes–quality, privacy, and security—are part of the review of any DCT.
Privacy
Security

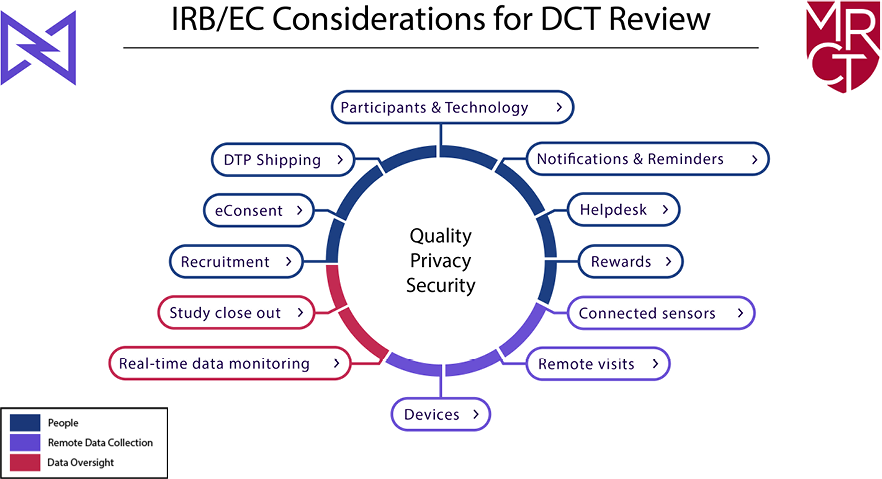
Figure 1: IRB/EC Considerations for DCTs
This document is organized by first describing the cross-cutting themes, followed by IRB/EC considerations for each DCT element, organized by the three domains. The recommendations include a summary of ethical issues associated with each decentralized approach, as well as a checklist that IRBs/ECs can use in their reviews. A comprehensive DCT checklist is also included at the end for reviewers who prefer to start with the full checklist and use the recommendations as a reference document. Although the guide is intended to assist those conducting ethical reviews of clinical trials, other stakeholders, such as sites and trial sponsors, can use these materials as part of clinical trial planning and to interact more effectively and efficiently with IRBs/ECs.
Recommendations
Cross-Cutting Themes
Quality, privacy, and security
As with all trials, consideration must be given to participant privacy and confidentiality, whether the DCT involves a tele-health visit, remote capture of data, use of a device, or delivery of an investigational product to the home. Many privacy risks differ fundamentally in quality and nature from traditional clinical trials. In addition, DCTs must ensure that data quality and data security are preserved, regardless of the nature of data collection, transfer, storage, and use.
Participant considerations
DCTs may use technology and devices to recruit participants and obtain consent for participation (i.e., informed consent.) As with all trials, recruitment, consent, and enrollment must be conducted in a manner that promotes understanding and allows participants to make informed decisions. However, IRBs must also consider issues of equal access, ability to use technology, and adequate infrastructure to support the use of the technology or provide remote encounters. If participants are to be recruited through social media, for example, there are extra steps that need to be taken to protect people’s privacy. Additionally, there are considerations for notifications and rewards and for shipping devices to participants. A helpdesk is often useful to help participants resolve technology-specific issues or to refer them to appropriate, licensed clinical personnel for issues related to the trial protocol. Each of these issues is considered in turn.
Remote Data Collection
In a DCT, data can be collected leveraging technology or via patient care provided outside of a research site. Because participants may have remote visits in their homes or at a local clinical site or pharmacy, special care must be taken to protect their privacy. Devices (such as tablets or smartphones) can either be provisioned by the participant (bring-your-own-device) or provided by the investigator or sponsor. In either case, care must be taken to ensure equitable, private access to devices and related infrastructure for data transfer, storage, and access. The same is true for connected sensors (such as spirometers and continuous real-time glucose monitors) that are connected to the internet. Additionally, the connected sensor may be an already approved device or commercially available sensor designed to collect outcome information specific to the trial or itself may be the device under investigation. In the latter case, different ethical and regulatory considerations arise.
Data oversight
Although healthcare providers may have the capability to monitor data continuously and in real-time in a DCT, data will likely be monitored at specified intervals. This information should be communicated to participants along with what information (if any) will trigger action from the study team. For study close-out, processes may be a bit more complicated than with a traditional trial, and remote devices will need to be collected, destroyed, or wiped of information,
The use of technology and the ability to conduct aspects of a trial remotely in a DCT has great potential, but there are many special considerations for the adequate protection of the participant and their data. This guide is intended to help IRBs/ECs understand the considerations and inform decisions about oversight.
Quality, privacy, and security
Quality, participant privacy, and security are areas that may require particular attention throughout a DCT regardless of which decentralized approaches are used.
Quality.
“Quality” in clinical trials is defined as the absence of errors that matter to decision-making—that is, errors that have a meaningful impact on the safety of trial participants or the credibility of the results (and thereby the care of future patients). In the context of an ethical review of a decentralized element, the IRB/EC focuses on both data and safety aspects. Safety considerations are broad and include everything from remote reporting of (and acting upon) adverse events to assuring the ICF appropriately discloses all information potential participants need to know relative to aspects of a trial that will be decentralized. In addition, because DCTs are often used to reach more participants through technology and local/mobile providers, issues related to equity, inclusiveness, and diversity are often more pronounced and require the examination of specific ethical questions. Similarly, data collected through remote approaches (including sensors, tech apps, and local providers) raise data accuracy and completeness questions, as well as questions about the validation of the technology used. A protocol developed through a quality-by-design approach, and which comprehensively describes the decentralized elements of a trial, facilitates a more efficient and effective ethical review of a DCT by an IRB/EC.
Privacy.
Participant privacy is one of the key facets of an IRB/EC review in any clinical trial. In a trial using decentralized approaches, there are unique ethical considerations associated with a specific technology (e.g., telehealth, apps, and sensors) and with the providers who interact with participants in their homes or other locations away from the trial site. In essence, a DCT interposes one or more intermediaries in the relationship between a principal investigator (PI) or the site staff and a participant. By doing so, new parties become privy to private patient information (e.g., personal identifiable information (PII), geolocation, activity, etc.) Accordingly, an IRB/EC may need to review additional contract terms, processes, and potential vulnerabilities of participants’ private information, as well as participants’ knowledge of who will have access, how their private information may be used, and what choices they can make about these things.
Security.
In a DCT, clinical trial data is collected outside of a traditional site. There are a number of regulations and guidances that address the collection, transfer, and submission of data in clinical trials for regulatory submission, e.g,, 21 CFR part 11. From an ethical perspective, IRBs/ECs should understand how the security of data collected through apps, sensors, and local/mobile health care will be assured, where the information will be stored, who can retrieve the data, and what parameters are in place to securely transfer the data to the PI and the clinical trial record.
PEOPLE
Clinical trial recruitment through social media
Introduction
Recruitment through social media can be conducted passively through advertisements, posts, or flyers or actively where researchers interact with individuals on a social media platform. In either case, the IRBs/ECs should be provided with information describing the approach and plans to obtain consent and documentation of consent activity from participants.
Advice for Investigators/Sponsors Proposing to Recruit via Social Media
- Provide the IRB with a statement describing the proposed social media recruitment techniques, including:
- A list of all channels/websites to be deployed in recruitment.
- A script of the information, images, and format to be displayed or posted. (See information about eConsent content.)
- Describe recruitment methods:
- Passive recruitment involves the distribution of recruitment materials (e.g., ads, posters, flyers) with the aim of attracting potential participants to contact the research team for more information and consideration of enrollment.
- Provide all information, notices, and/or advertisements that are proposed to be posted about the planned clinical research. Include where such information will be posted.
- Describe whether and how this recruitment differs from the placement of a notice in a newspaper, on a bus, or on other public site as might be done in a traditional clinical trial.
- Active recruitment involves the interaction of research staff members with specific individuals with the aim of enrolling them in research.
- Describe how research staff will identify themselves when entering and while on the social media site and upon engaging any specific individual
- Attest that private personal and health information will not be retained, scraped, or stored from the site without consent.
- Describe how potential participants will be identified and approached, and how their privacy and confidentiality will be maintained.
- Describe the proposed elements of any planned conversation or interaction with potential participants.
- Attest that screening for participation or exchange of private personal and health information will not be performed on the social media site.
- Passive recruitment involves the distribution of recruitment materials (e.g., ads, posters, flyers) with the aim of attracting potential participants to contact the research team for more information and consideration of enrollment.
- Provide the IRB/EC with a statement certifying compliance (or lack of non-compliance) with the policies and terms of use of relevant websites.
- If the proposed techniques conflict with relevant website policies and Terms of Use
- Provide the IRB/EC with a written statement describing the apparent compelling circumstances that might justify IRB/EC approval, despite non-compliance with the website’s Terms of Use.
- If the IRB/EC requires the website to provide an exception from its policies or terms of use, the PI should provide the IRB with written documentation of the exception, if granted.
- Ensure that the proposed recruitment strategy respects all relevant ethical norms.
- Proposed recruitment should not involve
- Deception or fabrication of online identities.
- Members of the research team ‘lurking’ or ‘creeping’ social media sites in ways that members are unaware.
- Advancements or contact that could identify, embarrass, or stigmatize potential participants.
- Recruitment strategies should generally be designed such that:
- The reach of the post is purchased rather than based on “likes,” “sharing,” “retweets,” etc. since such behaviors might expose medical information unnecessarily to the extended network of the potential participant.
- Any promotion of the link will transfer the potential participant away from the social media platform and to a website from which information is not shared or at risk of being shared with one’s network.
- Trials are accurately represented in recruitment overtures.
- Proposed recruitment should not involve
- Potential trial participants found on social media are likely to engage on social media with some regularity. Therefore, a formal communication plan is needed for managing social media activities among enrolled participants, including:
- Steps to educate participants about the importance of blinding and how certain communications can jeopardize the scientific validity of a study (e.g., a informational section in the orientation or consent form)
- Triggers for intervention from the research team (e.g., misinformation or speculation among participants on social media that could lead to un-blinding)
- Interventions from the research team (e.g., corrections of misinformation or reminders about the importance of blinding on social media.)
- Consider making the posts only visible to group members to avoid medical information being shared (or imputed) when potential participants interact with the post.
- Ensure that the social media recruitment strategy complies with applicable country, federal and state laws.
Electronic Consent (eConsent)
Introduction
The goal of the informed consent process is for prospective participants to make a voluntary, informed decision about clinical trial participation. The use of eConsent digitizes the informed consent form (ICF), which allows for, but does not require, interactive enhancements, such as videos, hyperlinks to additional information about elements of the study, and quizzes or other self-assessments of understanding, intended to increase participant understanding and decrease participant and site burden. It also allows for the consent process itself to be virtual or remote.
Considerations for IRBs and ECs
- Description of trial risk:
- Trial risk does not impact the IRB/EC review of the informed consent process, which should enable participant understanding. The informed consent process may be problematic regardless of the trial risk.
- Trial risk does impact the nature of protocol review and consent, including trials using eConsent. For example, tele-visit or in-person assistance may not be required during the eConsent process for minimal-risk studies.
- Method for verification of participant (or Legally Authorized Representative [LAR]) identity:
- Comply with standards to verify individuals; these can vary by country, state, and institution. This process should not place an undue burden on the participant.
- If the investigator has no previous contact with or knowledge of the participant, more in-depth verification may be required.
- eConsent content:
- The language of the eConsent should be reviewed similarly to traditional consent.
- The IRB should pay particular attention to visual images, videos, and other information associated with the eConsent to ensure that:
- The information is fair and balanced and, as for all trials, not biased in favor of one arm of the trial, the intervention, or participation itself.
- Embedded hyperlinks should be reviewed to ensure that the information is fair and balanced, and non-promotional.
- The information in the eConsent is consistent with the rest of the informed consent and study.
- The information is accessible and/or alternative formats for the same or similar information is available.
- Translations of the eConsent, if available, are consistent.
- The information is fair and balanced and, as for all trials, not biased in favor of one arm of the trial, the intervention, or participation itself.
- Images are inclusive and considerate of the intended study population and community.
- IRB/EC review of the consent language may be performed while the platform is designed and developed. After the platform is developed, an expedited review can be completed. A working link can be provided for evaluation of the actual eConsent experience which potential trial participants would see.
- Appropriate features of eConsent should include:
- Ability to move forward and backward within the document.
- Ability to stop and resume at a later timeA method to verify identity as the participant or legally authorized representative (LAR).
- Translation to the preferred language of the participant.
- Modifications to the eConsent document and process for people with disabilities.
- Any included hyperlinks work.
- Possible inclusion of questions to assess ongoing understanding (optional).
- Short form eConsents are generally not acceptable.
- Necessary terms are defined in plain language for all consent forms. There may be additional terms to be defined in a DCT.
- Amethod for obtaining a validated participant signature must be provided.
- Assistance that will be offered to potential participants:
- If eConsent is fully remote:
- Participants need
- Access to technical assistance, if necessary.
- A method to receive help for questions regarding the trial or content of eConsent
- A printed or electronic copy of the signed eConsent.
- Participants need
- If eConsent involves a remote or tele-visit or is done in person:
- The responsible person(s) for assisting in the eConsent process should be identified.
- Any tele-visit conducted as part of the consent process should be documented.
- If eConsent is fully remote:
- Measures implemented to assure platform security:
- Documentation of platform security should be provided. Third-party vendors have often undergone review and have the necessary documents to show compliance with security regulations.
- If this is not the case, an IRB may require institutional validation of security or accept PI assurance that platform security is compliant.
- Storage of eConsent records:
- Investigators and sponsors should have or have access to:
- An audit trail to identify the participant, study staff, and the date/time of eSignature and PDF creation of an IRB-approved informed consent document.
- Access to the system that is restricted to appropriate personnel
- Appropriate archiving capability with restricted access and with all versions easily retrieved.
- Other questions to consider
- Where will eConsent records be stored?
- Will any vendors or 3rd parties have access to the records?
- Investigators and sponsors should have or have access to:
- Implementation of eSignature:
- The acceptability of eSignatures varies by country. The signature process should comply with national and local laws and regulations.
- When eSignatures are acceptable, the use of a Qualified eSignature, which entails providing a participant with a unique code that must be entered along with their eSignature, is considered best practice. However, this process is often burdensome, and the need for it must be considered in the context of a specific trial.
- Pediatric participants:
- The Parent/Guardian/LAR’s credentials should be used to create, verify, and document consent and assent in the eConsent system.
- Assent should be captured electronically as a separate field in the eConsent system.
Technology Use by Participants
Introduction
Participants enrolled in a DCT will likely interact with one or more technologies that facilitate consent, communication, and/or data collection. The processes and materials developed for the use of these technologies in a DCT should enable participant use, support participant access and inclusiveness, and ensure safety and data quality. IRBs/ECs should consider asking the below questions for a DCT trial where participants will use technologies.
Questions for IRBs/ECs to Consider
- Equitable access to the study:
- How will eligible participants access the technology that is required to participate in the study?
- Will the participant be expected to provide their own technology (device, software, app, and/or other)?
- Will the different sources of technology used by different participants impact data quality?
- If so, will a common technology be provided by the sponsor or investigator?
- Is the technology accessible for people with disabilities?
- Have alternative provisions for study participation be made, if necessary?
- In locations that do not have access to needed infrastructure such as broadband internet, will participants have an alternative option to participate in the study?
- What costs will the participant incur associated with trial technology?
- If costs are study-specific costs, will they be paid by the sponsor or investigator?
- Will reimbursement or an allowance for data plans be provided?
- Device Considerations – see recommendations related to Devices in DCTs, Direct-to-Participant Shipping, and Help Desk documents
- Participant Education, Resources, and Training
- Technology allows for easy linkages to various types of information (training videos etc). In the study being reviewed, what types of information (e.g., disease or condition, local providers) are available to participants through the technology?
- Did the IRB review and approve the content of any hyperlinks in the consent and other participant-facing materials?
- If local providers are engaged, does the IRB/EC have appropriate oversight?
- If the IRB/EC does not have oversight, what risks should be considered and how will they be managed?
- Are participants directed to any promotional materials, advertisements, or commercial resources?
- Have linked websites been included in the IRB protocol for review and approval?
- Will any of these sites collect, store, or transfer personal information?
- Are study-specific materials (see eConsent tool) provided to participants via remote technologies? If so, does the process include a knowledge check?
- What instructions do participants receive regarding how to use the devices and technology in the study?
- Is the PI or someone from the study team available to participants during the trial if questions or issues arise?
- Is contact information for the PI or study staff, or a helpdesk resource, (see helpdesk tool) available outside of the technology itself? Note: while this issue is not unique to DCTs, it is often lost in the context of DCTs.
- Are materials easy to read and in plain language?
- Are materials translated into the preferred language of the participant?
- Will the translations be available via technology and/or paper?
- Will paper copies of the educational resources and training materials be available?
- Technology allows for easy linkages to various types of information (training videos etc). In the study being reviewed, what types of information (e.g., disease or condition, local providers) are available to participants through the technology?
- Privacy/Confidentiality considerations
- Does the technology include the collection of protected health information (PHI), PII, or sensitive information?
- Are there measures in place to assure privacy/confidentiality (e.g., end-to-end encryption, secure servers, two-factor authentication)?
- Is direct contact with participants limited to non-sensitive messages?
- Is remote removal of all stored information from the technology (e.g., remote wiping of the device) possible in cases of device loss or theft?
- Special Populations – Prisoners
- Because of limitations on access to devices, the internet, and other systems, DCTs may not be feasible in prisons
- If a trial participant is sentenced to jail or prison after enrolling in the study, the study team must determine whether it is feasible to continue that person’s participation in the trial. If feasible, the IRB/EC should be notified; in certain jurisdictions, the IRB/EC may require re-review of the protocol to ensure conformance with local requirements (e.g., 45 CFR 46 Subpart C in the US.)
- Special Populations – Children
- If the study involves children, is the technology appropriate for children? Has the parent or guardian approved its use?
- Are there specific regulations that address studies involving children?
- In the US, if the study involves children ≤ 13 years of age, does the study comply with the Children’s Online Privacy Protection Act of 1998, 15 U.S.C. 6501–6505 as amended?
- Are the instructions related to the use of technology age-appropriate?
- Will the child be able to access information using the device or technology other than study-specific materials?
- Does the parent or guardian understand that information outside of the study can be accessed?
- Are there any devices that will be used by the participant’s parent or guardian? Are there any instructions for the participant’s parent or guardian regarding any devices being used in the trial?
Notifications and Reminders
Introduction
A component of many DCTs is the ability of the participant and study team to communicate with one another in real-time. In these trials, the site investigator or study team can create and send both automatic and on-demand notifications and reminders to the participant to help increase the completion of study procedures in a timely fashion. Participants themselves may appreciate the receipt of electronic notifications and reminders because they decrease the burden of “having to remember” their responsibilities, among other benefits. Digital engagement platforms typically include systems for sending notifications and reminders. Even in the absence of specific DCT platforms, SMS texts can be sent to the participant’s mobile phone, although the financial cost to the participant should be recognized and compensated. How these notifications and reminders are delivered—the frequency, tone, method, etc.—should be optimized to improve the participant’s clinical trial experience as well as trial outcomes. Often, the participant can choose how and when to receive the communication (see below), thus increasing engagement and satisfaction, although whether participant will be allowed choice may depend upon the study itself. There are also concerns to address, including those relating to participant confidentiality. Finally, the IRB/EC should consider whether the device is provided by the participant, investigator, or sponsor.
Participant Preferences
As much as possible, digital engagement platforms should be implemented and/or communications (e.g., through a personal mobile phone) should be configured to facilitate participant choice. For example, participants should be allowed to choose:
- Whether to receive the communication as a text, email, or phone call
- When to receive the communication, including frequency, timing, and, if applicable, triggers (e.g., notification if continuous glucose monitoring indicates an action is needed)
- Language preference for communication
- For example, in the case of a reminder to take a study medication, having chosen to receive this reminder as a text message, the participant could determine:
- Preference for English or another language
- Timing of notification, whether 15 minutes before, 5 minutes before, and/or at time study medication is due
- One, two, or more notifications for the same event
- Whether a participant action (e.g., acknowledgment via a “like”) is required to turn off reminder:
- For example, in the case of a reminder to take a study medication, having chosen to receive this reminder as a text message, the participant could determine:
Note: whether, when, and how communications are received and responded to, and whether the communications are required, optional or flexible will often depend upon whether they are an integral part of the research protocol or of safety monitoring.
Risk Mitigation
There are confidentiality concerns with notifications and reminders. The smartphone or other electronic device on which notifications are displayed may not be private and may be shared with others. Even if the device is private, it may be lent to or held by another person, or a third parties may view information on the screen, either intentionally or unintentionally. The information contained within the communication may reveal or imply some private information Of course, the device may be ‘hacked’ maliciously or stolen. There may be data security concerns with the data platform itself. Mitigation strategies include:
- Minimize information in notifications, with specific attention to sensitive or potentially sensitive information. While apps can generally be designed so the text of a notification is only shown when the smartphone is unlocked, this protection is more difficult with SMS because the settings are controlled by the smartphone user.
- Provide information through an app that requires personal log-in information to access (note: this approach may reduce the likelihood that participants will see the notification or reminder, e.g., if they fail to log in regularly)
- Establish an understanding with the participant about intended uses and potential risks of the device, its ownership, and access in advance
- Provide a dedicated device, if necessary
- Require two-factor authentication, when necessary
- Limit direct messaging to non-sensitive information, when possible
- Provide for a remote wipe of the device in cases of stolen or potentially hacked devices
- Assess platform data security measures for compliance with institutional and internal policies and requirements.
There is also the possibility that participants will stop paying attention to, or ignore, the communication. The participant may be annoyed by the disturbance of constant reminders and notifications and may even “turn off” or disable the communication. The communication may be blocked or otherwise not delivered unbeknownst to the participant and/or study team
While these are not specific to DCTs, there are somewhat different and heightened concerns in DCTs. Mitigation strategies include:
- Solicit periodic feedback from the participant regarding experience
- Intermittent remote monitoring (e.g., determine if notifications are received and/or read)
- Create a feedback path such that the participant must respond to communication and any failure to respond will alert the study team
- Modify the content or form of the communication periodically
- Use an incentive reward system if necessary (see Rewards document)
Questions for IRB/ECs to Consider
- Is the implementation of notifications or reminders likely to create an undue burden or introduce risks to privacy or confidentiality?
- Does the communication contain potentially sensitive or identifiable information? Can the inclusion of that information be further minimized?
- Are there data security concerns introduced by these communications and, if so, can they be eliminated?
- What control does the participant have over the form, frequency, and content of the communication (e.g., can the participant turn off reminders that are critical for safety?)
- What is the plan if the participant fails to respond to a safety-related notification?
- What are the consequences, risks, and potential harms:
- If the communication fails?
- If the communication is received by a third party?
- Is there a monitoring plan for the system of notifications and reminders? A monitoring plan is necessary if there are meaningful consequences to the failure of notifications. Note, however, that in a particular trial, smartphone medication reminders might be just a convenience or redundant with other reminders that are in place. If the study team determines that missing an occasional dose is unlikely to cause harm to participants or create a data quality issue, a monitoring plan may not be necessary.
- Does the study include participant feedback, and is it elective?
Direct-to-Participant Shipping
Introduction
Some DCTs involve shipping a research product (e.g., drug, investigational medicinal product (IMP), diagnostic, device, or sensor), directly to the trial participant. There are a variety of considerations for the IRB/EC regarding direct-to-participant shipping, including country, state and local laws, regulations, and considerations specific to the product, including its route of administration, stability, safety, and accountability for the product. In addition, whether the research product is shipped by the investigator, sponsor, or third party will impact the IRB/IEC review, given that participants’ personal information will be collected. All this information is generally included in the protocol document reviewed by the IRB/EC. IRBs/ECs evaluating protocols involving Direct-to-Participant shipping should consider the following questions for each item being shipped.
Questions for IRB/ECs to Consider
- What is the item being shipped?
- Is there a description of the device, sensor, drug, or IMP being shipped, including technical specifications and safety data sheets, included in the protocol or attached materials?
- Have any unique shipment considerations such as temperature sensitivity, fragility status, protection from light, safety during shipment and receipt been considered?
- For an investigational medicinal product (IMP):
- If an IMP is being shipped, are the requirements of Good Manufacturing Practice (GMP) and Good Distribution Practice (GDP) for the IMP fulfilled?
- Are reconstitution fluids and procedures and/or devices for product infusion or administration included, if applicable?
- Are appropriate measures to maintain blinding of the trial in place, if applicable?
- Where will the IMP be shipped, and what interstate or international laws and regulations must be adhered to?
- What storage conditions at the home of the trial participant are required?
- Who is accountable for IMP and responsible documentation? If the participant is accountable, what education, assistance, and oversight will be provided? Consider:
- Shipment receipt
- IMP storage
- IMP distribution
- IMP return or alternative disposition of the unused product
- How will the participant be adequately informed of the purpose of the IMP, its risks, and proper use?
- Who will administer IMP to the participant?
- Does a healthcare practitioner need to assist with training or the initial administration(s)?
- Is a healthcare practitioner necessary to mitigate risks of acute significant adverse events?
- Who will monitor participant use?
- How will adverse events be identified, reported, and treated?
- Are mechanisms that permit rapid identification of the IMP in the event of a medical emergency in place, accessible, and communicated?
- How is compliance monitored?
- Who will administer IMP to the participant?
- What are the participant safety concerns, including receipt, handling, storage, administration, and adverse events of IMP?
- For devices or sensors
- What training is required for the participant to use the device or sensor appropriately?
- What further cost or burden will the participant bear (e.g., WIFI, data)?
- Except in rare circumstances, there should be no penalties for damage or loss.
- How will items be labeled and shipped?
- Have shipping procedures and the proposed delivery plan been optimized for conditions specific to the item being shipped (e.g., device, sensor, drug)? For example, shipping of temperature-sensitive drugs should include means (1) to monitor temperature, (2) to determine if the temperature threshold is exceeded, and (3) to ensure someone is present to receive and adequately store shipment at the destination when delivered.
- Does the protocol describe how to label boxes? Does the labeling allow participants to identify the shipment quickly as part of the trial while also balancing privacy concerns? Are necessary measures in place to ensure that participants’ personal data are protected from unauthorized or accidental disclosure?
- Are there instructions for the shipper, such as not to leave the package with neighbors?
- What is the plan to verify arrival?
- Does the protocol include plans to verify delivery to the correct participant?
- If a shipping error occurs, is there a contingency plan in place to replace IMP, device, or sensor quickly? This plan is essential for IMPs and time- or temperature-sensitive drugs
- Is participant training required to use the item being shipped? How will assistance be given, if needed?
- Will the trial participant be given appropriate information and agree to share the personal data necessary for the item to be processed, shipped, received, and used?
- Does the trial participant need instruction in advance about the correct storage and use of the shipped item? If training is required, is a training plan and methods to ensure comprehension included in the protocol?
- Has the burden on participants been minimized for direct-to-participant shipping?
- Is the burden different than anticipated were the IMP distributed at the research site?
- Are there additional participant risks by direct-to-participant shipping compared with traditional trials?
- Do participants need 24/7 access to either assistance about the item being shipped or the protocol? If not, will lack of aid possibly cause significant risks to patient safety or data quality?
- Do participants have contact information for either technical or protocol-specific assistance?
- Will the plan for direct-to-participant shipping be a barrier for specific participant populations or participants in certain locations?
- Are hybrid solutions possible (e.g., direct-to-participant shipping, shipping to a local healthcare facility or pharmacy, etc.)?
- Optimally, participants can choose how they wish to receive the IMP. If direct-to-participant shipping is necessary, then informed consent should explain that shipment to their personal address is required.
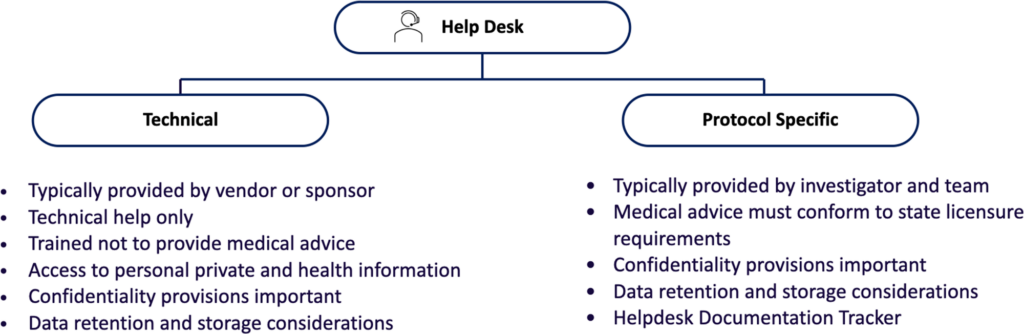
Helpdesk Function
Introduction
A “Helpdesk” function is important for DCTs because it helps individuals respond to, manage, organize, and “troubleshoot” software or hardware issues that arise in technology-enabled tasks. Notably, many participants or their caregivers may not be technically savvy, familiar with the mobile health (mHealth) technology utilized, or even with core functionalities such as access to the internet. Thus, a Helpdesk is typically either a customer service team or a technical system that provides technical support and assistance to users of computer technology. Helpdesks also usually have a ticketing and reporting system for insight into repetitive problems and as a guide to necessary improvements, updates, and ‘bug fixes.’
In addition to software and hardware questions, participants sometimes contact the Helpdesk with questions about the clinical trial, medical questions, symptoms, or other issues. Because it is not the intent of the Helpdesk to answer these questions, participants are referred to the study team by the Helpdesk (see below). However, even in these routine interactions, personal health or other personally identifiable information may be disclosed. Thus, the Helpdesk team must be trained appropriately, and, if the service is provided by a vendor or outside entity, appropriate protections of confidentiality and privacy must be established. Optimally, the vendor will be a Business Associate of the institution conducting the research.
The Helpdesk is NOT the destination for medical advice; all emergencies and medical questions should be referred promptly to appropriate personnel and documented. Participants should be advised that, in general, the Helpdesk will guide the participant to the correct person to answer the question or provide a solution, including being transferred to qualified medical personnel.
Note that it is important to consider site and participant (user) support holistically. While the Helpdesk is the primary touchpoint to support users once an issue has been discovered, there are a variety of methods to empower users and mitigate issues prior to Helpdesk contact. The “best” Helpdesk call is the call that never happens because the system is easy to use, training has been effective, and “self-help” modalities, should a problem arise, are available.
Considerations for IRB/EC Review
- Based on the type of question, separate people or teams should respond. Helpdesk personnel should have appropriate qualifications, training, and delegation for their role in the study.
- Calls by participants to the Helpdesk should be logged, but no record linking the identity of the caller to the study, condition, or sensitive data should be maintained unless necessary, and if necessary, appropriate procedures to maintain confidentiality and privacy should be in place.
- Call logs, tracking, and reports should be audited for retention of personal health or identical information.
- The IRB/EC can always ask to review the Helpdesk standard operating procedures, personnel training and qualifications, and other materials if needed.
Questions for IRB/ECs to Consider
- Will trial participants have 24/7 access to Helpdesk support?
- If not, how likely will the lack of assistance cause meaningful risks to patient safety or data quality?
- Will, and how will, the helpdesk staff triage participant questions?
- Will the telephone voice prompt system or operator triage calls be based on the type and nature of the participant’s question?
- Does the Helpdesk have scripted questions that are routinely asked?
- If so, the questions and format should be reviewed.
- Will identifying personal information be maintained securely and only retained when necessary (e.g., for follow-up)?
- Will all helpdesk staff, whether internal or external to the organization, be appropriately trained?
- Will the Helpdesk accommodate underserved and underrepresented populations?
- Will calls be answered in the preferred language of the caller?
- If not, how will translation and/or interpreter services be provided?
- Are teletypewriter (TTY) and assistive devices available to accommodate people with disabilities?
- Will calls be answered in the preferred language of the caller?
Additional Considerations Regarding Helpdesks
When designing a Helpdesk for a trial or if IRBs/ECs have concerns about answers to the above questions, it may be helpful for sponsors and researchers to review these considerations and best practices.
- First contact resolution.
- The more likely a Helpdesk is to resolve a user’s issue in the first contact, the more likely a user is to remain in the study. Complex issues or an ineffective Helpdesk result in user frustration that may drive site friction and patient dropout.
- Study complexity.
- The more complex a study (number of endpoints, measures and instruments, number of customizations, etc.), the less likely it is that a helpdesk can effectively troubleshoot a user’s issue in a timely manner or on first contact.
- Helpdesk training.
- The effectiveness and regularity with which a helpdesk is trained on both the “product” (technical) and its use in the study, and appropriate referral for study specifics (protocol), the higher likelihood of successful issue resolution.
- Adequate provisions for maintaining the confidentiality and privacy of callers.
- Provisions for confidentiality and privacy should be established and documented.
- In the US, if the institution is a HIPAA-covered entity, then it may be appropriate for vendors and call centers to execute a business associate agreement or other contract with the institution.
- Both the vendor and Helpdesk staff should be trained in:
- Confidentiality
- Implicit bias and cultural humility
- Customer service
- Accommodating different needs if the participants (e.g., slowing down and simplifying information for people who have difficulty with technology).
- All records, call logs, and communications should be transferred to the investigator and/or sponsor, stored securely, and retained per record retention policy.
- Provisions for confidentiality and privacy should be established and documented.
- Sponsors and sites should aim for a positive communication experience for the participant and/or their caregivers.
- Scripted questions should be asked to identify whether the caller has a technical, medical, or protocol-based question.
- Based on the type of question, separate people or teams should respond
- Technical staff should be trained not to respond to medical, research, or protocol-specific questions
- If access to personal health or identifiable information, do not maintain a record linking the caller to the study, condition, or sensitive data.
- Call logs, tracking, and reports should be audited for the retention of personal health or identifiable information.
- If there is a medical question or emergency, the caller should be referred to medically responsible personnel, health care providers, or emergency services, as necessary.
- Medical and professional licensure requirements must be met if medical advice is provided.
- If there is a research or protocol-specific question, qualified research staff should be positioned to answer, familiar with the protocol.
- Medical and professional licensure may be necessary depending on the advice given.
- The role of the Helpdesk personnel should be understood. If the Helpdesk personnel are engaged in medical advice or research processes, they should have appropriate qualifications and delegation for the role in the study.
- The investigator and sponsor should be informed about technical issues raised by the participants and should consider whether the technical issues are impacting trial participation, data quality, or execution of the clinical trial protocol.
- Technical issues should be fixed in a timely manner, and the investigator and sponsor may need or wish to review any time delays in issue resolution.
- The Helpdesk should provide a report on issues encountered and how they were resolved.
Rewards for Participation
Introduction
Research participants are often remunerated for their participation, and may be reimbursed for expenses, compensated for their time and burden of participation, and offered incentives for their continued engagement. The IRB/EC is responsible for assessing that remuneration is proportionate to the burdens of research participation and that it does not present an undue influence on research participation. There are parallels in non-DCT research that have been discussed extensively in the literature, so the focus of this document will be on aspects of rewards that differ in the DCT context from more traditional research. Note that, like traditional trials, studies vary in the extent of participant burden. A study adding eConsent is not fundamentally different from a study with paper consent. However, studies with a number of expectations for at-home participant responsibility (e.g., wearables, sensors, medication self-administration, at-home surveys, telemedicine, or fully virtual trials) may increase trial burden or even shift the burden from sites to participants. Financial compensation should reflect the time, burden, and expectations of the participant, and the IRB /EC should consider the offer of payment very similarly to that considered in traditional trials.
The nature of rewards in DCTs can, however, extend beyond financial payments. When other types of rewards are introduced, specifically through a digital or mobile platform, those rewards often come in the form of gamification and acquisition of “rewards” for completing a task or entering additional data. The participant can get “caught up” in the game, whether because it is fun (or even compelling) or because of the promise of the reward. Further, participants may not, in the moment, consider whether certain data may be sensitive or private. While IRBs/ECs typically evaluate risks and benefits—and do so in the absence of any consideration of compensation—digital reward systems are often behavioral in nature, and therefore may be both more effective at impacting participant behavior.
Questions for IRBs/ECs to Consider
The IRB/EC should be provided with a summary of how the incentive system is designed, including: what rewards are offered; what requirements must be met in order to receive each reward; and what limits, if any, are in place that restrict the rewards that can be earned (e.g., daily reward limits) and/or restrict the amount of time that participants can dedicate to earning rewards (e.g., maximum hours per day that can be spent while still receiving rewards).
- Do risks to privacy, confidentiality, data storage and transfer change if rewards are given in DCTs? If so, how?
- Will the participant consider the risks and benefits of sharing personal data differently through mobile platforms, tele-visits, or visiting nurses compared to how participants would consider the risks and benefits during in-person visits and via paper forms?
- Will providing rewards in the planned fashion change the willingness of the participant to share their personal data in ways that create undue risk of harm?
- Are there any rewards or features of a reward program that are of particular concern (e.g., because they are engineered to introduce addictive behavioral patterns that have high risk of leading to serious interference with daily life among the target patient population for the trial)?
- Can the participant engage in choosing the goals of the reward?
- Are rewards earned through persistent engagement with technology or software?
- Do rewards accrue evenly or is the schedule of rewards modified by the responses of the participant? In other words, the participant receives X points for answering questions about themselves, but 3X points for answering questions about their sexuality.
- Are there concerns for persons with addiction or addictive personalities?
- Are there additional concerns for children who are participating in a DCT trial and are given access to different types of devices?
- Can the promise of the gift of the device and/or software be considered “undue influence,” impacting the voluntariness of participation in the study?
- Have any conditions of the reward been considered? For instance, if the gift is earned only at the completion of the trial, will that inappropriately influence the participant’s decision to withdraw? Is the reward dependent on sharing specific information?
- Will depreciation affect the value of the device and/or software over the course of the research?
- Is the value of the gift of the device or software proportionate to the time and burden of the research and appropriate for the protocol?
- What provisions have been made if the device is lost during the trial? (See tool for Devices for additional considerations)
- Is there any functionality, information, or other feature that should be removed, disabled, or modified prior to transfer to the participant as a reward?
As in other aspects of DCTs, particular attention to access, transfer, use, disclosure, and safe storage of personal information is necessary.
Specific considerations for participation rewards in DCTs
DCTs are often, but not always, enabled by electronic communications and mobile technologies, and that may be the most salient difference from traditional trials. In the context of “rewards,” DCTs offer the possibility of several qualitatively different kinds of rewards.
First, there is access to the sensor (e.g., fitness tracker, smartwatch, continuous glucose monitoring system) itself, if the sensor is provisioned for the research rather than provided or brought by the participant. In advance of the research, the study team should decide, and the IRB should review, whether and under what conditions the sensor will remain with the participant at the end of the trial, and what to do in the event of sensor loss during the study. Access to software drives similar considerations.
Another difference in DCTs is the use of gamification as an incentive, which may increase participant engagement. Gamification strategies refer to the use of gaming techniques to motivate (“nudge”) certain behaviors. Those behaviors are often positive (e.g., the participant is tasked with an increasing step count each day), but a concern arises when the “game” can only be “won” (or points accrued) if the participant agrees to do or disclose something that they would not have otherwise chosen to do or disclose (and if what they do or disclose is problematic). For example, it would be problematic if the only (or easiest) way to advance or accrue points in the game was by disclosing contact information of friends or certain social activities, information that the person would not have disclosed but for the reward. IRBs should ensure whether gamification is only nudging or renders the clinical trial into a game in itself. Gamification should not mislead participants, who should understand that they are a part of a trial (not a game). Rewards should incentivize people to disclose the information necessary to complete the outcome measures of the research as opposed to “as much information as possible.”
Gamification is used throughout the tech industry and is not distinctly different in the DCT context, except that the IRB/EC has a chance to review the testing and rewards. For IRBs/ECs reviewing gamification approaches, there are different risks and vulnerabilities to be considered, in addition to those of the research itself. People with addiction or addictive behaviors may be at greater risk of being “drawn into” the game or app, depending on the circumstances, and addictive behaviors are not typically considered in eligibility criteria. Digital rewards can also have a higher perceived value for the target audience than someone outside of that target audience could reasonably anticipate. Representative community input may be helpful in this evaluation.
REMOTE DATA COLLECTION
Remote Visits: Including Telemedicine, In-Home Visits, and Local Providers
Introduction
With DCTs, interactions between the participant and study team can be either virtual, using telemedicine or video-enabled visits or conducted by local providers who can accommodate seeing the participant at a nearby clinic or laboratory center or visit the participant in their home. “Bringing the trial to the patient,” when a site or hospital visit is not necessary, decreases the burden and possibly the expense of participation and is generally appreciated by participants. Some participants, however, prefer to visit the trial site for personal reasons (e.g., a reluctance to invite someone into their home, lack of privacy) and, therefore, whenever there is a choice as to the method of visits, the participant should be offered that choice. Here we enumerate considerations for DCT visits enabled through telemedicine (including both tele-visits (e.g., telephone only and video visits)), in-home visits, and local providers.
Telemedicine
Telemedicine involves one or more virtual visits between patient and investigator or research team member up to and including a completely virtual relationship between patient and research study team and/or the use of a digital platform in the absence of a physical (hands-on) exam to maintain notes and progress reports and to record clinical and personal observations. In addition, telemedicine visits may be accompanied by local laboratory or imaging capabilities, if necessary.
The potential benefits of telemedicine include:
- Convenience for both participants and sites because it reduces time, effort, and possibly expense
- Increased geographic reach (e.g., rural settings) and diversity (e.g., race/ethnicity, age) of study populations
- Greater participant flexibility
- Decreased burden on participants and researchers by reducing travel
- Preservation of social distancing with decreased exposure in case of infection, epidemic, or pandemic
- Decreased spread of disease and time research team spends with infected patients
- Potential to increase participant access, enrollment, and retention
Policies, coverage and payment rates, technologies, and familiarity with services provided by telemedicine are continuing to evolve. The same is true with knowledge and experience with telemedicine in clinical research. Telemedicine decreases the time, expense, and inconvenience of the ‘visit’ for participants, while it increases access to medicine in remote and rural areas. Further, the number of physiological sensor technologies (e.g., smartphones, blood pressure cuffs, pulse oximeters, activity trackers, glucometers, and scales) is increasing.
Challenges to the adoption of telemedicine
- Inequality
- Internet access, connectivity, and access to and comfort with the use of digital technologies varies among different demographic and geographic populations (“digital divide”). The problem may disproportionately affect people of color, Indigenous peoples, people with disabilities, older adults, low-income households, and people who live in rural areas. For example, the cost of data access plans must be considered, may be limiting, and may be a disincentive for participants who have limited cellular plans.
- Inclusion of people with disabilities in research may be more challenging.
- Translations
- Software and software platforms may not be available in multiple languages, limiting participation for individuals whose preferred language is other than English.
- There is limited availability of multiple languages for software and platforms
- Confidentiality
- Some individuals may not have private or isolated areas in their homes to host a telemedicine visit or may be unable to disable digital monitoring systems such as Alexa or Google Assistant. Disclosure to third parties may occur unintentionally; this risk must be mitigated. For example, participants may be able to conduct a telemedicine visit at a local church or pharmacy that has a private space where a participant may have a remote visit.
- Security
- Like all remote technologies, security must be addressed for all data, including data collection, transfer, storage, and retrieval related to the video.
- Validation
- How data will be validated must be addressed before leveraging remote technologies. The sponsor or investigator, for example, might send a participant a fitness tracker to monitor exercise and heart rate or ask a participant to send a picture of their feet on a scale to verify the weight.
- The identity of participants must be verified and identify clinical site personnel must be confirmed and documented.
- Professional licensing
- In advance of enabling telemedicine visits, professional licensing for investigators, if needed, must be considered, as well as state and country telemedicine laws.
- In the US, for clinical trials of a drug or device where there is a telehealth or video-conferencing visit, the investigator generally needs to be licensed in the state where the participant is located. Internationally, telemedicine is even more challenging.
- In advance of enabling telemedicine visits, professional licensing for investigators, if needed, must be considered, as well as state and country telemedicine laws.
- Variable reimbursement for a visit, if allowable and applicable
- Sharing of clinically relevant study data with physicians involved in the care of the patient.
- Costs of implementation
- Lack of clarity in and/or conflicting regulations and applicable laws
- Security and delivery concerns when delivering prescribed medication or devices (see investigational products)
- Providing adequate PI oversight may be challenged if contract (non-study) personnel are engaged.
In telemedicine visits, IRBs/ECs should consider:
- Participant preferences for a method of interaction with the research study team
- The nature of the study interventions, if any, that will be performed during a telehealth or video-conferencing visit and whether there is a need for someone else to be present?
- Privacy considerations for the participant. Is there a private setting for the telemedicine visit?
- There should be detailed information to the participant that telemedicine visits may expose private information, and that creating a private setting for these ‘visits’ are preferred.
- The provider and study personnel should ensure they do not have any protected health information visible on the screen if they are in their office.
- Equitable access to the internet, cellular data, devices, software, technical support
- The languages available in software and software platforms
- Modifications and accommodations for the inclusion of people with disabilities
- Data privacy and security in collection, transfer, and storage
- Methods of data validation and remote monitoring
- Shipment, receipt, administration, and disposal of investigational and other research products and their documentation
- The ability of the PI to provide adequate oversight of healthcare providers (HCPs) recruited for study activities.
In-Home Visits
In-home visits involve a research study team member, their designee, or a third-party vendor visit the participant’s home, usually because face-to-face interaction is necessary or preferred. Visits can be necessary due to required medication administration; collection of specimens, observational data, or other data; or safety concerns for the participant, including adverse events. An in-home visit is convenient for the participant and decreases their time, effort, and costs of visiting the study site; the research team incurs costs and time investment. Participants have expressed concerns about personal visits to the home regarding of privacy. For example, there may be neighborhood concern or stigma involved when a gowned and masked person arrives at a home, or the inability to find a private or isolated area in the home away from other people.
Other issues also need to be understood, such as: is the professional visiting considered an “investigator” on the trial? Do they have appropriate credentials, licensure, and training? Are they handling the investigational product(s)? Is adequate emergency care available, if needed? Is the research team member aware of mandatory reporting requirements (e.g., child or elder abuse)? Is there continuity in research staff performing home visits to engender trust and communication between participants and research team members? Will the PI be able to provide adequate oversight of trial conduct?
In summary, with in-home visits:
- The research study team either directly performs in-home visits or contracts with third-party vendors (with sponsors or study team directly) to conduct face-to-face visits and interactions with participants
- IRBs/ECs must consider whether third-party vendors are engaged in the research and whether the training, qualifications, and credentials of the personnel involved must be verified and/or if reliance agreements and/or other contractual agreements to define responsibilities and liability are needed.
- Travel burden is eliminated (e.g., time, expense, inconvenience) for participants when providers visit their home; this must be weighed against potential new burdens (e.g., privacy, safety, scheduling challenges. Options (e.g., for the site or in-home visits) should be provided whenever possible.
Potential benefits
- Convenient for the participant in time, effort, and expense (decreased burden)
- Greater flexibility for participants
- Helpful for studies that require face-to-face contact and when participants prefer not to or cannot enter or travel to a clinical setting/site
- Licensed professionals can administer treatments at and during home visits
- Home-visiting professionals can directly observe and interact, collect specimens, and ascertain patient-reported data
- Allows for a larger and more diverse study population
- Reduced opportunity costs for participants (time, expenses of traveling and/or childcare)
Potential Challenges
- In some particular social circumstances for the participant, in-home visits may not be convenient or appropriate
- Time and expense costs might be greater for the research site
- Potential privacy and confidentiality concerns of participants with home access
- Potential increased burden on patients (perception that house needs to be clean)
- Regulatory and administrative issues with third-party vendor contracting (e.g., credentialing, licensure, accountability, liability)
- Challenges to PI oversight of contracted parties or use of non-study personnel
- Regulatory uncertainties (e.g., who is engaged? In the US, who is an investigator requiring an FDA Form 1572 or Delegation of Authority?)
- Different capacity for and time to emergency care (i.e., call 911 versus crash cart on site)
- Challenges when specialized equipment, significant testing, or procedures are required
- Concerns for maintaining data security and transfer among the study team, participants, and third-party vendors
- Security and accountability concerns when delivering prescribed medication or devices
For in-home visits, IRBs/ECs should consider:
- Participant preferences for the method of interaction with the research study team
- Risks to privacy and confidentiality of participant
- Availability of emergency and other care
- Status of the healthcare provider: competencies, licensure, status as the investigator or engaged research personnel (e.g., on FDA Form 1572)
- Responsibility grid for providers and third-party vendors if engaged
- Mechanisms to ensure adequate PI oversight
Local Visits
For “local visits,” a participant may visit a local provider, laboratory, imaging center, or other site for their required research visit. Laboratory tests and imaging studies can often be performed locally, and the results sent to the responsible investigator. This is convenient, particularly in settings where routine labs or imaging are needed, and if there is equivalence between results obtained at the primary versus the local site. A local visit can also be used when face-to-face evaluation or specialized testing must be conducted in a clinical setting. Unless the services rendered are considered standard of care, the local site or must have appropriate training (e.g., GCP) and instruction.
In summary, with local visits:
- The study requires the participant to have contact with a clinician
- Participants are seen by local providers (possibly their customary providers), laboratories, and imaging centers
- The research study team or sponsor may contract with third-party vendors in local communities for patients to visit
- Data are transferred to the investigator study site
- Decreases, but does not eliminate, travel burden and opportunity costs to patients
Potential benefits
- Permits face-to-face visual and physical assessment of participant
- Less burden (e.g., travel distance)
- Relationship with local provider preserved
- Emergency services available
- More in-depth evaluations of the patient
- Accurate and validated study data
Potential Challenges
- Requires some travel, time, and expense for participants
- Training of local providers must be provided and assured
- Regulatory issues and responsibility for third-party vendors (credentialing, licensure, accountability, status)
- Monitoring/regulation of protocols at third-party local providers
- The local provider may or may not be an investigator
For local visits, IRBs/ECs should consider:
- The need for a face-to-face participant visit, laboratory tests, or imaging study
- Whether the local site or provider is adequately trained, credentialed, licensed, and familiar with the specific research protocol
- Assurance of PI oversight assured
- Data security and privacy for data collection, transfer, and storage
- Responsibility grid for providers and third-party vendors, if engaged
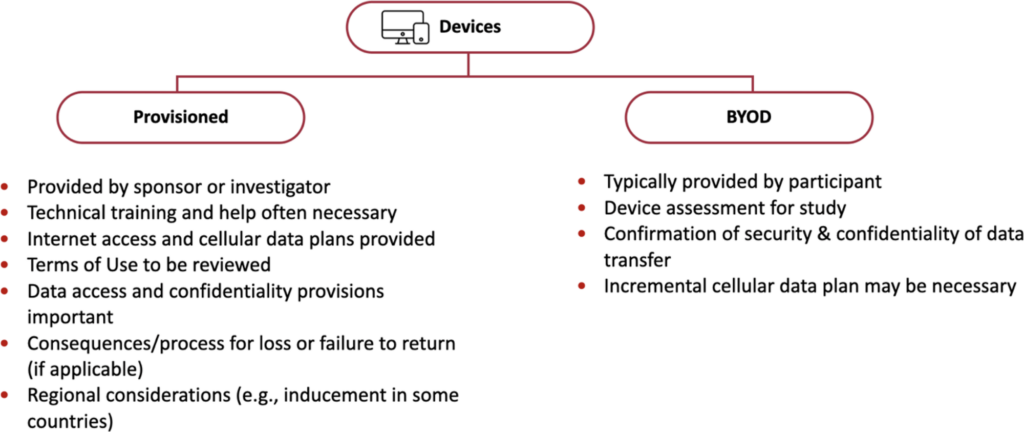
Devices (smartphones and tablets) in DCTs
Introduction
Smartphones and tablets (here, collectively termed “devices”) can be configured either as Bring Your Own Device (BYOD) or provisioned devices. They are often used in DCTs to help participants, investigators, sites, and sponsors collect data. Devices can be vehicles for participant data collection through electronic patient-reported outcomes (ePROs), observer-reported outcomes (ObsRO), and other eCOAs (electronic clinical outcome assessment). These assessments are usually deployed using applications downloaded onto the device.
Note that some studies involve investigation of the application or software itself, which raises questions of risk and benefit for the IRB/EC. For instance, a study may involve an app for the assessment of cognition. While this app may be part of a DCT and loaded onto a device, the IRB/EC will wish to review additional potential safety and other issues than those described here.
This document covers only non-investigational devices (smartphones and tablets) devices as objects in common use, deployed for the purpose of conducting the DCT; for information about wearables, connected devices, and applications, see the Connected Sensors section.
IRBs should understand which types of devices (provisioned or BYOD) are being used.
Differences between categories of Mobile Devices
BYOD devices
Although some sponsors are hesitant to allow participants to bring their own device (BYOD), some participants may prefer to use their own device in a clinical trial. BYOD can offer benefits such as improved data collection as owners are generally comfortable and experienced using their own devices.
Considerations for IRBs
- Equity and equitable access
- For example, if a smartphone is required, for those who do not have their own device will one be provided? Does that decision have implications for the equitable selection of participants?
- Provisioned devices ought to be considered if the BYOD device or data plan/Wi-Fi availability is inadequate. The decision to provide devices to participants unable to BYOD helps to ensure equity, as everyone would have equal opportunity and access.
- A minimal set of requirements for the BYOD hardware and software should be predefined, and a clear list of such requirements, written in plain language, should be made available to the investigator, study team, and participant. The requirements should define the compatible make/models and operating systems of devices.
- Sponsors should consider how to update provisioned and BYOD devices to maintain current versions of software.
- Each participant’s device should be assessed to ensure that the BYOD device meets the minimum device requirements to run the trial apps. Older models, for example, may no longer be supported or may not work well for this purpose.
- The cellular data plan must accommodate data collection and transfer needs in the trial or be augmented by the study team or sponsor. If the data plan will not be reimbursed, the informed consent process and document should alert prospective participants to the likely costs that will be incurred.
- Security and confidentiality for data transfer should be ascertained and confirmed, with particular attention to sensitive or health information.
- It should be understood if the application accesses information from other applications on the device and if there are risks associated with that additional information.
- Informed consent considerations for BYOD:
- Costs associated with data plans/WIFI essential to trial data submission must be disclosed. Supplemental data plans should be provided if needed.
- Trial participants must consent to the use of their own device during the clinical trial.
Provisioned devices
Provisioned devices are devices provided by the sponsor or institution, and provide a more controlled, standardized data capture platform than BYOD. Thus, investigators and sponsors should define whether all participants will be given a provisioned device or only those who do not have an adequate personal device along with the implications this may have for participant selection.
Considerations for IRBs/ECs
- The IRB/EC should review the plan for reporting and replacing a device lost, damaged or stolen. While rare exceptions may exist, the participants should not incur penalties for losing provisioned devices. Particular attention should be paid to any proposed penalty (e.g., covering replacement costs, withdrawal from the study) that will be imposed upon the participant.
- Any change to the study to add a penalty or cost for a lost, damaged, or stolen device should be considered a change to the research and require IRB/EC review and approval.
- Sponsors and/or investigators should ensure that a provisioned device can be tracked, and that the tracking functionality is active only upon notification of loss.
- Sponsors and/or investigators should ensure PHI and clinical trial data are encrypted at rest and in transit and be able to lock and wipe the device remotely.
- If a device is lost or stolen, the device should be remotely accessed and wiped to remove all information.
- The provision of a replacement device need not be reported to the IRB/EC.
- Any possibility of disclosure of PHI through lost or stolen data should be reported to the IRB/EC and to any covered entities as a potential HIPAA violation, as applicable.
- The IRB/EC should anticipate that the type of provisioned device will vary by region.
- Data collected and transmitted automatically should be available to both the investigator and the sponsor (i.e., to verify that data was not modified)
- Informed consent for provisioned devices should include the following information:
- Costs associated with associated data plans, if any.
- Instructions in the event that a device is lost, damaged, or stolen.
- Information on who will provide replacement device(s) and limitations in the number of replacement devices, as applicable
- Description of the process a trial participant will follow if a device is lost, damaged, or stolen. Optimally, there should be no penalties for lost, damaged, or stolen devices. If any penalties (e.g., financial reimbursement, removal from study) are proposed, the penalty should be explained in the informed consent process and documented in the consent form. Penalties should be non-punitive and reasonable based on the depreciated value of the device.
- Disposition of the device at the end of the study:
- Whether the device be returned to the sponsor and the method for return, such as participant return to study site or pre-paid return shipping label provided.
- Whether the participant can keep the de-commissioned device at the end of trial. If the participant retains the device, depreciation of the device should be calculated and documented so that the participant incurs no tax consequence.
Device logistics and cost
- Sponsors rarely handle the logistics of their own devices, and either buy the device outright or lease it from a technology partner.
- When the device is a required tool for trial participation, participants should be provided with appropriately administered and documented instructions to use the device properly.
- Trial participants should have access to a Helpdesk (see helpdesk tool) device troubleshooting.
- Cost considerations:
- IRBs/ECs may wish to consider the cost of the device in their deliberations, particularly if possession of the device may subject the participant to risk of harm (e.g., in some settings, possession of the device will expose the person to privacy risks or risk of theft).
- IRBs/ECs may wish to consider situations where the participants are allowed to retain the device after the trial, and whether the possibility of retention is an appropriate incentive to participate in the trial. In that context, the depreciated value of the device at the end of the trial should be considered.
Connected Sensors
Introduction
Connected sensors include technology products such as wearables, sensors, and other connected devices (e.g., spirometers and continuous real-time glucose monitors) that can be connected to the internet. These mobile sensors capture and process data using algorithms to generate measures of behavioral and /or physiological function.
Note: this section should be used in conjunction with the section on Devices (smartphones and tablets).
Questions for IRBs/ECs to Consider
- Is an investigational testing authorization by the health regulatory authority required for the sensor? (In the US: is the study exempt from investigational device exception (IDE) regulations?)
- What is the purpose of the sensor being used?
- How is the device classified?
- Is the sensor an investigational medical device in the study?
- For instance, in the US, is it a Significant Risk (SR) or Nonsignificant Risk Medical (NSR) device?
- Is the sensor validated for the purpose and population for which it is being used?
- What data is the sensor collecting?
- If the sensor is being used to monitor an at-risk participant group, consider:
- How often will the study team review sensor data?
- Will the study team be able to intervene in a timely way if abnormal vital signs or findings are detected by the sensor?
- Note: data that may trigger the need for timely intervention may relate to blood pressure, heart rate or rhythm, or other vital functions, but may also relate to other types of data such as suicidality, depression, etc
- Where is sensor data stored?
- If a third-party vendor is involved in studies using connected sensors, does the vendor have access to the data?
- Is any potential re-use by the vendor defined and clearly stated in the informed consent document?
- Is such re-use compliant with relevant legislation and requirements for ethical approval?
- Is the third-party vendor a business associate or contracting party required to maintain privacy and confidentiality? If not, what provisions are in place for protecting participant privacy and confidentiality?
- Are there additional applicable contractual agreements that should be considered?
- What, if any, burden does the sensor impose on the participant?
- If the sensor is required for trial participation, are participants provided with appropriately administered and documented instructions to properly use the sensor?
- Do trial participants need access to a 24/7 call center to troubleshoot issues with the sensor as they arise? If not, how likely will the lack of assistance cause meaningful risks to patient safety or data quality?
- Will the sensor be collected at the end of the study?
- Optimally, the value of the sensor will be depreciated over the course of the study, and sensor return will not be required. If return is not required, will the sensor be cleaned or wiped to remove personal research data?
- Are there processes in place that make sensor return as simple as possible for participants?
- While rare exceptions are possible, there should be no financial or other penalties for lost or broken sensors.
REMOTE DATA COLLECTION
Real-time Data Monitoring in DCTs
Introduction
Once a DCT study is launched, study sponsors and sites must monitor several aspects of the trial on an on-going basis to ensure its proper execution, data integrity, and participant safety. While on-going monitoring is an expectation of all clinical research, in the context of DCTs, it may involve dynamic “real-time” monitoring in addition to new technologies that can be deployed. This document focuses on continuous monitoring of real-time data and the opportunities and potential challenges presented by these digital technologies.
Study elements that can be monitored frequently or in real-time
- Enrollment
- Participants’ completion of required tasks and scheduled activities
- Participant requests to the Helpdesk and the study team
- Notification schedules and responses to those notifications if enabled and required
- Sensor data, e-Patient Reported Outcomes, and other outcome measures
- Safety signals
- Endpoints and outcome measures
- Investigator and sponsor reports
- Overall compliance
Not all of these study elements are unique to DCTs, but some require special considerations. Here we focus on the issues that are either unique or more pronounced in DCTs than in traditional, site-based trials. For example, more frequent (or real-time) monitoring of participants’ compliance with required tasks.
Questions IRBs/ECs to Consider
- What type of study data are available to the investigators and to the participants?
- Are these data unique in some way?
- Will access to data or results impact trial integrity or blinding?
- Are there additional safety concerns?
- Is there an expectation to respond to real-time reporting of adverse events? Are the mechanisms in place to assure timely response (e.g., 24hr monitoring)
- Are there confidentiality and/or privacy concerns?
- Are staff and systems in place to respond to real-time receipt of participant data, if necessary?
- Does the site/investigator have sufficient resources to support the DCT’s requirements for real-time monitoring?
Impact on the Participant
- Dynamic, real-time data or result return:
- Will the participant and/or treating physician/investigator have access to data or results during the trial?
- What type of data will participants have access to during the trial?
- Will this data be understandable and/or actionable by the participant?
- Will access to the data impact participant behavior, safety or study results?
- Will participant access to data influence future behavior or impact study blinding?
- Does the ICF clearly explain the extent to which data submitted by participants will be monitored by study staff?
- Participants may believe that study staff are monitoring their data in real-time, whether or not they are, and fail to alert study staff to possible adverse events or assume staff will respond to possible adverse events.
- Is the quality and burden of participant submission of adverse events considered?
- Has the protocol anticipated the accuracy, frequency, and burden of participant reporting of adverse events?
- Will reporting be systematic and comprehensive?
- Are systems in place to ensure that study staff will review and respond to reports in a timely manner?
- What procedures are in place for responding to serious or unanticipated adverse event?
- What approaches or mitigation strategies are in place to assure adequate participant compliance for completing questionnaires throughout the study? Participants may attenuate to and tire of completing questionnaires, increasing the potential for missing information.
- What information is communicated to participants about the study staff availability and response time? Are there specific considerations related to data collected through sensors or submitted by participants?
- Has the study team or sponsor minimized the data collection to only that necessary for the study outcomes? Has metadata collection also been minimized?
- What back-up systems are in place should devices, technologies, data transfer, or data storage techniques fail? Are back-up systems proportionally secure?
Study Closeout
Introduction
Study or trial closeout is the process by which all activities related to the clinical trial are reconciled, recorded, reported, and stored at the end of the trial. Study closeout is an important component of quality; properly executed, it will also simplify access to study documentation and data access in the future or if the study undergoes post-closure audit or inspection.
The responsibilities of investigators and their study teams, and of the IRB/EC in its oversight role, are similar in DCTs and traditional trials. The major focus of the IRB/EC should be on aspects that differ between the two types of trials, including the disposition of the device (if not provided by the participant) and research data, and reconciliation and disposition of any investigational product, particularly if the product has been delivered to a home or local sites.
Given the nature of participant engagement in decentralized clinical trials, some investigators prefer to manage the closeout process with each participant as they complete their study participation, and then later closeout the overall study itself. We first consider activities related to the participant, and second, related to the entire study, with an emphasis on those activities relevant DCTs. There are different processes for the collection, transfer, and storage of data from DCTs than traditional trials.
Study Closeout Activities Related to Individual Participants
The following end-of-study considerations should be reviewed:
- The participant has completed their role in study, including all study procedures and data collection.
- Participant data have been captured and source documentation is available and verified.
- Data is complete, clean, and the database is locked.
- All study data have been transferred from devices.
- Participant has returned devices and sensors, or instructions for destruction provided if required.
- If participant used their own device (phone or tablet), provisions for deletion of study data (i.e., cookies and sensitive personal data) and apps have been communicated and/or verified.
- Only necessary personal identifying data have been retained in the study data
- Investigational product, if shipped to participant, has been accounted for, and any residual product has been returned and/or destroyed, as applicable.
Study Close-out
1. Data
- Only necessary personal identifying data have been retained in the study data
- All eCRF/EDCs have been completed and submitted as applicable
- All queries have been resolved
- Participant access has been removed or deactivated from the connected device or other eCOA data capture/patient engagement platforms
- Quality Control database
- Database lock
- Data storage arranged, retrieval and backup recorded
2. Investigational product or study drug and pharmacy:
- Site/Local/home investigational product or study drug accounted for
- Accountability logs and forms reviewed and filed
- Shipping logs documentation is reviewed and filed
- Any discrepancy documented, investigated, and resolved
- Pharmacy documentation is in order
- Remaining investigational product or study drug, materials, and compounds are returned or destroyed as outlined in the protocol or agreement with the sponsor
3. Documentation and Closeout
- All unanticipated problems, adverse events, and serious adverse events have been documented, resolved, and reported as required
- All study documentation available and securely retained
- eConsents or ICFs on file
- Study files reconciled with Trial Master File
- Study closure forms submitted to IRB/EC
- IRB/EC closure confirmed
- Any discrepancy documented, investigated, and resolved
- Final inventory/reconciliation of remaining or unused trial supplies, including devices loaned to participant and equipment loaned to the local site, completed
- Trial supplies returned to the sponsor or destroyed, as applicable
- Biospecimens recorded, stored for future use (where permitted), and documented
- Note: Do not close out the study if biological specimens containing individually identifiable information are maintained in the approved study-specific repository or upon which analysis or research continues. The study may be closed if biospecimens are transferred to a different repository that has separate IRB/EC approval.
Checklist for IRB Oversight of Decentralized Clinical Trials
This document is a checklist of the issues that IRBs/ECs should consider when reviewing a decentralized clinical trial. The checklist is organized into three domains: people, data collection, and remote data oversight. Within each domain are relevant decentralized approaches that may be part of a DCT and the questions for IRB/EC review. For additional information on the ethical considerations of any particular element, please consult the full set of recommendations.
PEOPLE
eConsent
- Determine the risk level of the trial (minimal/no minimal risk).
- Ensure the consent includes appropriate descriptions of DCT modules used, the functionality of eConsent platform, method of obtaining signatures, and assistance available to potential participants.
- Verify the identification method for participant and site staff identities (should be included in consent).
- Ensure the platform is secure and acceptable (e)Signature methods are used per region.
- Consider special considerations for vulnerable populations, including pediatric populations.
Social Media Recruitment
- Review approach and consent plans
- Consider how research staff are identified on social media platforms
- Review PHI storage and ensure use is appropriate and secure
- Ensure
- privacy of potential online participants is protected
- Social media channels use for recruiting are compliant with applicable regulations
- Management and communication plan is in place for social media actions of participants and consequences
Technology Use
- Ensure equitable digital access to the study.
- Review the device considerations and participant education materials, resources, and training.
- Consider privacy and confidentiality considerations.
- Consider special populations, such as prisoners and children.
Notifications and Reminder Checklist
- Is the implementation of notifications or reminders likely to create an undue burden or introduce risks to privacy or confidentiality?
- Does the communication contain potentially sensitive or identifiable information? Can the inclusion of that information be further minimized?
- Are there data security concerns introduced by these communications and, if so, can they be eliminated?
- What control does the participant have over the form, frequency, and content of the communication?
- What is the plan if the participant fails to respond to a safety-related notification?
- What are the consequences, risks, and potential harms:
- If the communication fails?
- If the communication is received by a third party?
- Is there a monitoring plan for the system of notifications and reminders?
- Does the study include participant feedback, and is it elective?
Direct to Participant Shipping Checklist
- Understand what is being shipped and associated shipping requirements
- Consider labeling and shipping
- Rapid identification of trial shipment balanced with privacy concerns
- Receipt verification procedures
- Contingencies for shipping errors
- Burden to participants
- Real time assistance protocol and or technical
- Consider equity issues: Does DTP create unforeseen barriers for certain populations
- If investigational medical product is shipped, ensure
- Compliance with good manufacturing & good distribution practices (GMP & GDP)
- All materials are included for administration
- Blinding can be maintained
- Adherence to local, national and international shipping laws for IMP
- A plan for receipt, storage, accountability and maintenance
- Plan for documentation
- Recipient training and readiness for receipt and administration of IMP
- Participant monitoring including AE reporting and responsibilities
- Rapid unblinding of IMP in medical emergencies procedures
- For devices and sensors, consider
- training/cost or penalties in case of loss
Help Desk Checklist
Assess study complexity when considering
- Is there 24/7 Access for participant related to questions about participant safety or data quality
- Ensure
- Appropriate Help Desk staffing and training (including training frequency)
- Appropriate triage of helpdesk issues and questions to medical protocol and technology resolution
- Systems for using helpdesk trends as way to assess technical issues that may impact participation and retention
- Scripts for different situations
- Appropriate PHI recording, maintenance, and discarding
- Accommodations of underserved and underrepresented populations
- Languages / TTY assistance/assistive devices for people with disabilities.
Rewards Checklist
- Consider risks to privacy, confidentiality, data storage and transfer change if rewards are given
- Ask if using technology for rewards administration, changes the risk perception to privacy/confidentiality/data storage & transfer
- Understand if there are any rewards or features of a reward program engineered to inculcate addictive behavioral patterns or to provide sensitive information
- Consider if there is undue influence in the device disposition plans at end of trial or are proportionate to the burden, and will not influence voluntary nature of participation
- Understand costs and/or penalties associated with loss of provisioned devices or sensors
- Ensure appropriate “data and tracking cleaning procedures” in place before transfer to the participant of the provisioned devices or sensors
- Consider effects on persons with addictions or children
Remote Data Collection
Remote, In-home, and Local Visit Checklist
| Considerations | Tele visit | In- home | Local provider |
|---|---|---|---|
| Consider whether access to internet, cellular data, devices, software, and technical support will be equitable across participants | |||
| Ensure multiplicity of language availability of software and software programs | |||
| Verify Modifications and accommodations for inclusion of people with disabilities exist | |||
| Verify Methods of data validation and remote monitoring | |||
| Shipment, receipt, administration and disposal of IMP and other research products | |||
| Ensure Participant preferences for method of interaction with research study team | |||
| Evaluate and Mitigate Risks to privacy and confidentiality of participant | |||
| Ensure Data privacy and security in collection, transfer and storage | |||
| Ability of PI to provide adequate oversight of HCPs recruited for study activities | |||
| Verify status of healthcare provider: competencies, licensure, protocol familiarity, Form 1572 | |||
| Verify Task log for providers and third-party vendors, consider if they are engaged in research | |||
| Ensure availability of emergency and other care if needed | |||
| The need for a face-to-face participant visit, laboratory tests, or imaging study |
Devices in DCTs Checklist
| Equity: Consider | BYOD | Provisioned |
|---|---|---|
| Equity issues when minimum device requirements are not met. | ||
| What the options are for participation if data plan is insufficient | ||
| Security and confidentiality in data transfers | ||
| Ensure Consent includes | ||
| Costs and use of device and whether replacements devices are available (at what cost) | ||
| ensure Instructions and process are available if device lost, damaged, stolen | ||
| Consider disposition at end of study (keep or return) | ||
| Ensure Appropriate instructions and training | ||
| Ensure access to helpdesk troubleshooting |
Connected Sensors Checklist
Consider
- Purpose of the sensor (critical vitals/at risk group)
- Regulatory status of sensors
- Frequency of monitoring of the sensor data
- Where sensor data is stored
- Burden of sensor to participants
- Clarity of where to get help for sensor issues
- Disposition of sensor
Real-Time Data Monitoring Checklist
Considerations for investigators and site staff
- What real time data is available to investigators?
- Are data unique in some way?
- Will real time data access unblind study?
- Issue related to confidentiality or privacy of data
- Are staff systems and resources in place to respond in real time
- Safety concerns related to real time monitoring expectations
Considerations for participants
- What real time data is available to participants?
- Will data unblind?
- Will access to data be understandable/ actionable?
- Will access to data impact participant behavior, safety, or study results?
- Is ICF language
- clear about what data will be monitored in real time, and
- clear about expectations for AE reporting to site staff?
- Participant burden on AE reporting
- Participant adherence to etasks (eCOAs)
- Resources to participants regarding staff availability and response times
- Data collection is fit for purpose and minimized
- Back-up systems are in place if technology fails to collect data
Study Close-Out Checklist
Data Considerations
- Consider completeness of source data and verification
- Ensure only required data retained in study records
- Ensure deactivation from provisioned devices/sensors/trial platforms and applications
- Verify Data storage privacy and security and all usual end-of-study activities
Investigational Medicinal Product and Direct-to-Participant Shipping :
- ensure accountability and documentation completed
BYOD
- Ensure BYOD Data transfer & removal (cookies, PHI, tracking) from participants BYOD/device
Provisioned device
- Ensure disposition of device is clear
Task Force Members
Barbara Bierer, MD, MRCT Center
Andrei Chiriac, MD, Medable
Leanne Madre, Medable
Evan Sohn, Harvard Catalyst
Pamela Tenaerts, MD, Medable
Darin Achilles, Food and Drug Administration
Alison Bond, Amgen
Megan Doyle, Amgen
Michelle Feige, Association for the Accreditation of Human Research Protection Programs, Inc
Jonathan Green, National Institutes of Health
Zachary Hallinan, Clinical Trials Transformation Initiative
Tara Isherwood, Syneos Health
Julie Kaneshiro, Office for Human Research Protections
Pat Larrabee, Rochester Clinical Research
Carole Legare, Health Canada
Craig Lipset, Decentralized Trials & Research Alliance
Nathan Morton, Coastal Carolinas Research
Lisa Murray, formerly MRCT Center
Frederik Grell Nørgaard, Danish Medicines Agency
Ann Meeker-O’Connell, Food and Drug Administration
Jane Perlmutter, Gemini Group
Jennifer Ribeiro, Bristol-Myers Squibb
Robert Romanchuk, Advarra
Leonard Sacks, Food and Drug Administration
Sana Shakour, University Michigan
Megan Kasmatis Singleton, Johns Hopkins Medical Institute
Joan Venticinque, Patient Advocate
Medable Subject matter experts
Andrew MacKinnon
Carl Franzetti
Florence Mowlem
Past
Andrew Johnson, formerly Medable, and
Lisa Murray, MRCT Center